Filtering¶
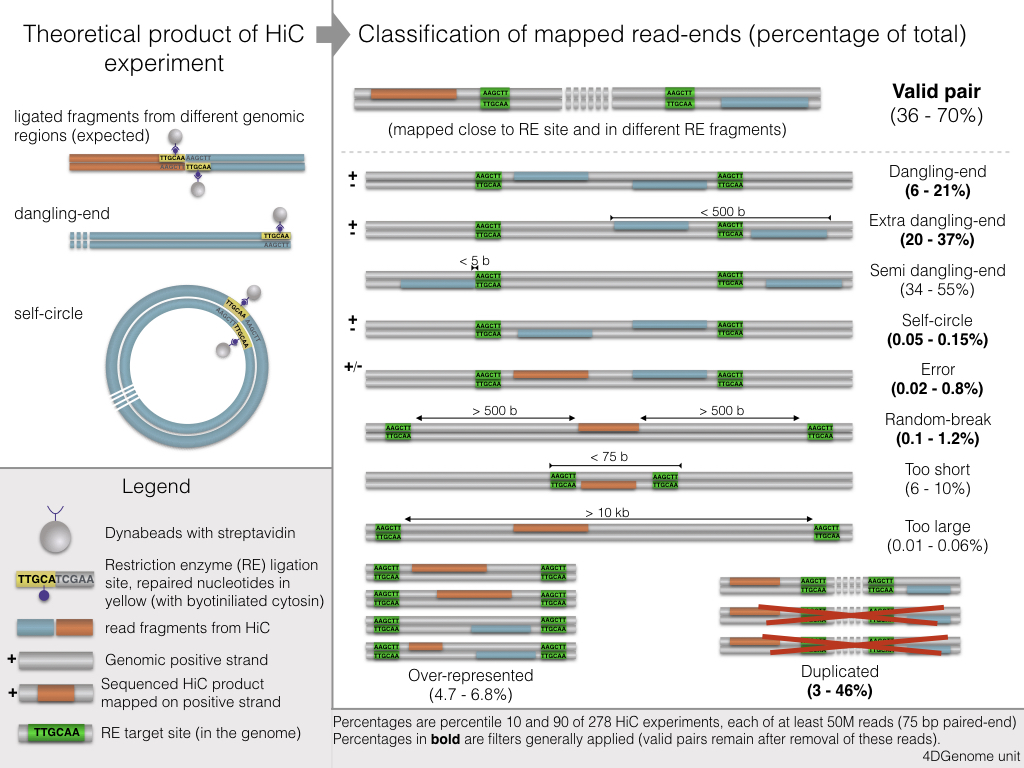
Filters¶
Pairs of mapped read-ends are filtered in order to keep only valid pairs. The filters available in TADbit are these one: 1. Self-circle: both read-ends are mapped to the same RE fragment in opposed orientation. 2. Dangling-end: both read-ends are mapped to the same RE fragment in facing orientation. 3. Error: both read-ends are mapped to the same RE fragment in the same orientation. 4. Extra dangling-end: the read-ends are mapped to different RE fragments in facing orientation, but are close enough (< max_molecule_length bp) from the RE cut-site to be considered part of adjacent RE fragments that were not separated by digestion. The max_molecule_length parameter can be inferred from the fragment_size function previously detailed. 5. Too close from RE sites (or semi-dangling-end): the start position of one of the read-end is too close (5 bp by default) from the RE cutting site. 6. Too short: one of the read-ends is mapped to RE fragments of less than 75bp. These are removed since there is ambiguity on where the read-end is mapped as it could also belong to any of the two neighboring RE fragments. 7. Too large: the read-ends are mapped to long RE fragments (default: 100 kb, P < 10-5 to occur in a randomized genome) and they likely represent poorly assembled or repetitive regions. 8. Over-represented: the read-ends coming from the top 0.5% most frequently detected RE fragments, they may represent PCR artefacts, random breaks, or genome assembly errors. 9. PCR artefacts or duplicated: the combination of the start positions, mapped length, and strands of both read-ends are identical. In this case, only one copy is kept. 10. Random breaks: the start position of one read-end is too far (> minimum_distance_to_RE) from the RE cut-site. These are produced most probably by non-canonical enzyme activity or by random physical breakage of the chromatin. Note, that to filter all these types of fragments the minimum_distance_to_RE parameter should be larger than the maximum_fragment_length.
cell = 'mouse_B' # or mouse_PSC
rep = 'rep1' # or rep2
Filter out dangling ends and self-circles¶
from pytadbit.mapping.filter import filter_reads
The max_molecule_length parameter used to filter-out
pseudo-dangling-ends can be extracted from the insert_size function
in previous section.
The min_distance_to_re, that affects the detection of random breaks,
should be large enough in order to contain almost all the fragments.
# this will last ~10 minutes
masked = filter_reads(
'results/fragment/{0}_{1}/03_filtering/reads12_{0}_{1}.tsv'.format(cell, rep),
max_molecule_length=750, over_represented=0.005, max_frag_size=100000,
min_frag_size=50, re_proximity=5, min_dist_to_re=1000)
Filtered reads (and percentage of total):
Mapped both : 87,862,102 (100.00%)
-----------------------------------------------------
1- self-circle : 87,761 ( 0.10%)
2- dangling-end : 4,813,620 ( 5.48%)
3- error : 18,165 ( 0.02%)
4- extra dangling-end : 12,933,038 ( 14.72%)
5- too close from RES : 21,372,275 ( 24.32%)
6- too short : 3,628,579 ( 4.13%)
7- too large : 1,256 ( 0.00%)
8- over-represented : 2,693,107 ( 3.07%)
9- duplicated : 1,605,185 ( 1.83%)
10- random breaks : 308,512 ( 0.35%)
This generates a dictionary with the different filters and the reads affected by each.
Apply filters on the data¶
from pytadbit.mapping.filter import apply_filter
apply_filter('results/fragment/{0}_{1}/03_filtering/reads12_{0}_{1}.tsv'.format(cell, rep),
'results/fragment/{0}_{1}/03_filtering/valid_reads12_{0}_{1}.tsv'.format(cell, rep), masked,
filters=[1, 2, 3, 4, 6, 7, 9, 10])
saving to file 66,612,427 reads without.
66612427
Evaluate changes¶
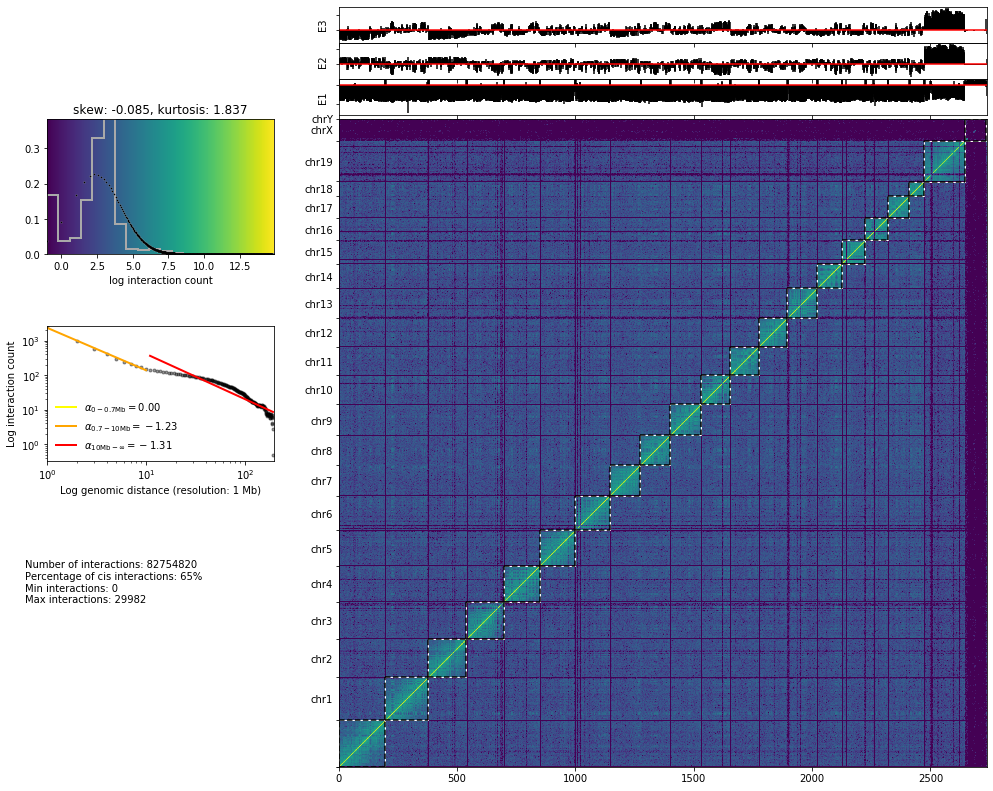
from pytadbit.mapping.analyze import hic_map
hic_map('results/fragment/{0}_{1}/03_filtering/valid_reads12_{0}_{1}.tsv'.format(cell, rep),
resolution=1000000, show=True, cmap='viridis')
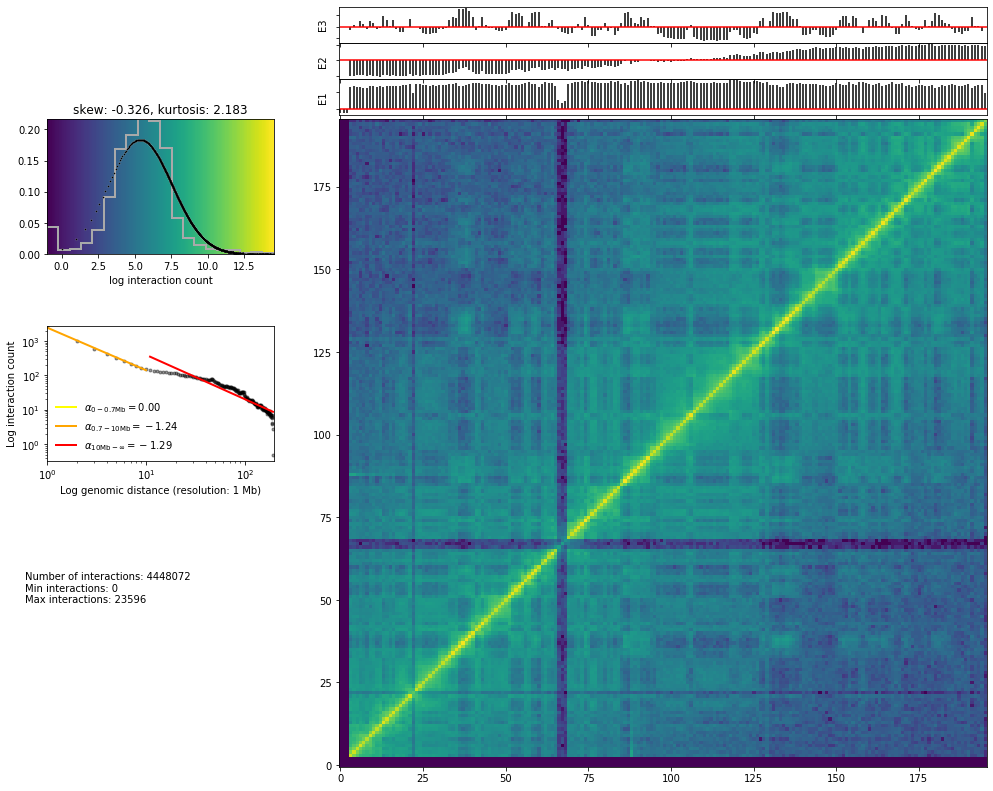
Zoom to a single chromosome or a region:
hic_map('results/fragment/{0}_{1}/03_filtering/valid_reads12_{0}_{1}.tsv'.format(cell, rep),
resolution=1000000, show=True, focus='chr1', cmap='viridis')
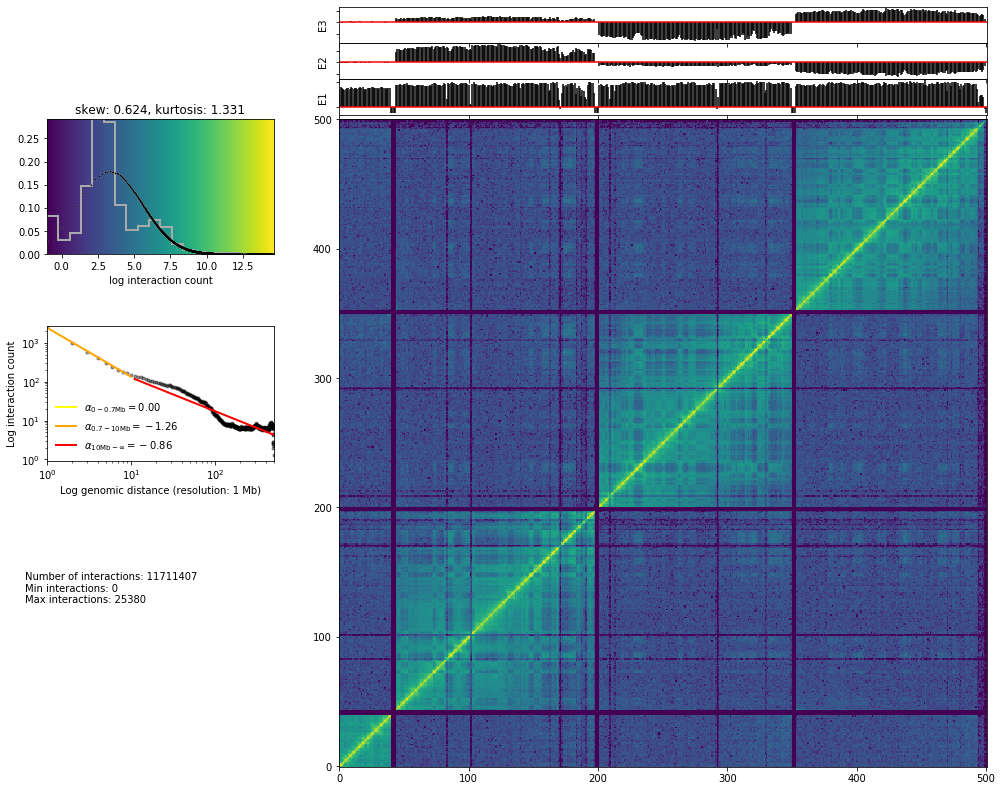
hic_map('results/fragment/{0}_{1}/03_filtering/valid_reads12_{0}_{1}.tsv'.format(cell, rep),
resolution=1000000, show=True, focus=(500, 1000), cmap='viridis')
Save to BAM¶
Working with TSV (tab-separated-value file format) files is very slow. For the next part of the tutorial we will be using BAM (binary-alignment-map) files, which are compressed and indexed.
Note: The fields we use in TADbit to generate a BAM file are not the conventional ones, we modify them as follows to store only the necessary information for the remaining part of the analysis: - Read ID (same as in the original FASTQ file) - Flag (binary mask for the application of the 10 filters previously described): 1. self-circle 2. dangling-end 3. error 4. extra dangling-end 5. too close from RES 6. too short 7. too large 8. over-represented 9. duplicated 10. random breaks 11. inter-chromosomal
For example if we want to keep only pairs of read-ends that are
excelusively inter-fragment contacts and that are not duplicated, we
would apply filters 1, 2, 3 (self-circle, dangling-ends, errors) and 9
(duplicated) resulting in a binary number like this: 00100000111 which
translates in decimal: 263. We could thus obtain these read-pairs with
samtools view -F 263. - Chromosome ID of the first read-end -
Genomic position of the first read-end - MAPQ set to 0 - Pseudo CIGAR
replaced by the mapped length of the first read-end, and information
about current copy (each pair is present twice in the BAM, P: first
copy, S: second copy) - Chromosome ID of the second read-end - Genomic
position of the second read-end - Mapped length of the second pair-end -
Nothing (*) (the field is usually reserved to sequence) - Nothing (*)
(the field is usually reserved to quality) - TC tag indicating single
(1) or multi contact (3 6 … number being the number of times a given
sequenced fragment is involved in a pairwise contact) - S1 and S2 tags
are the strand orientation of the left and right read-end
from pytadbit.parsers.hic_bam_parser import bed2D_to_BAMhic
bed2D_to_BAMhic('results/fragment/{0}_{1}/03_filtering/valid_reads12_{0}_{1}.tsv'.format(cell, rep),
valid=True, ncpus=8,
outbam='results/fragment/{0}_{1}/03_filtering/valid_reads12_{0}_{1}'.format(cell, rep),
frmt='mid', masked=None)
